|
|
V. 1.0 MIVEGEC-IRD / MAB-LIRMM-CNRS
|
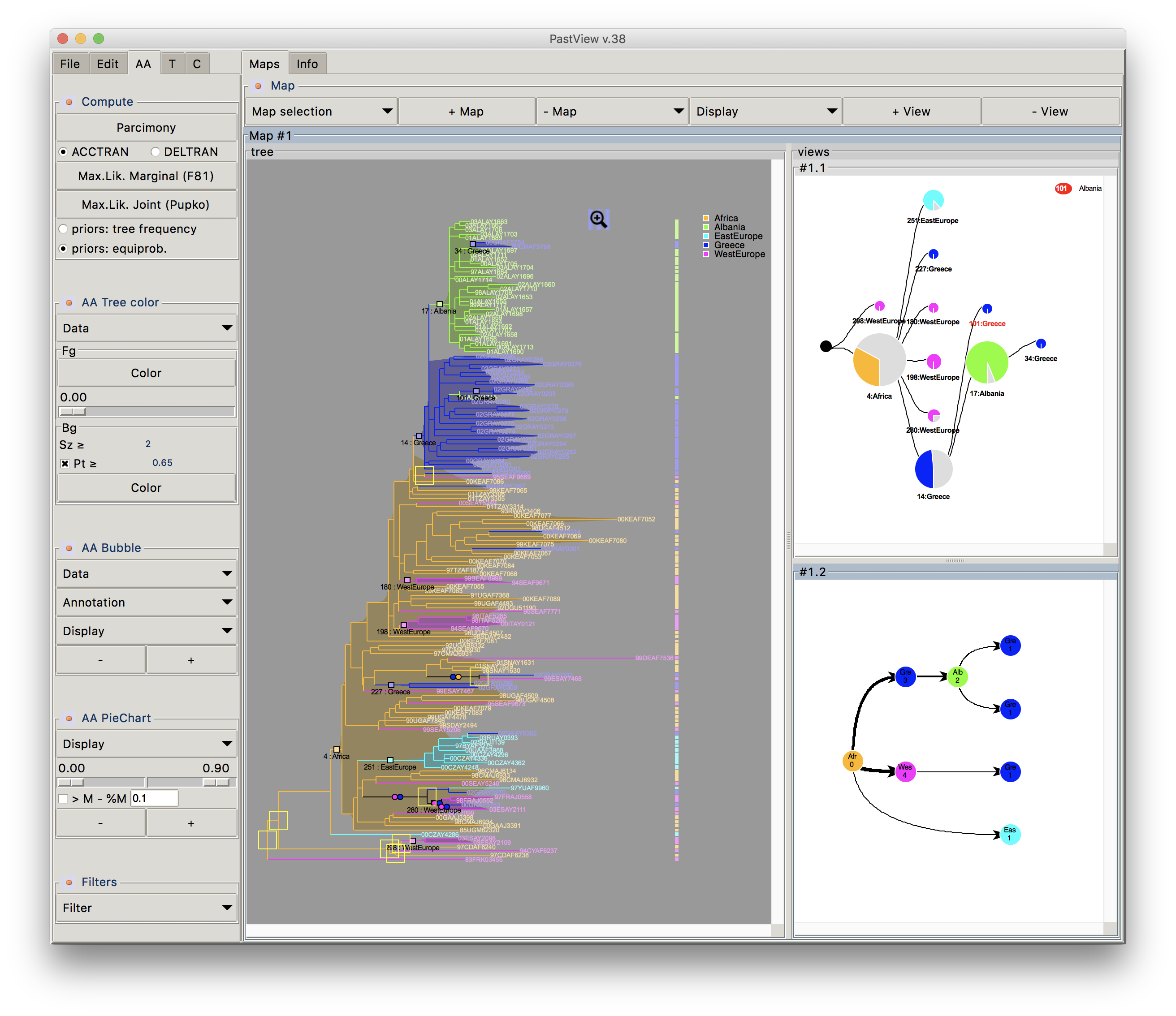
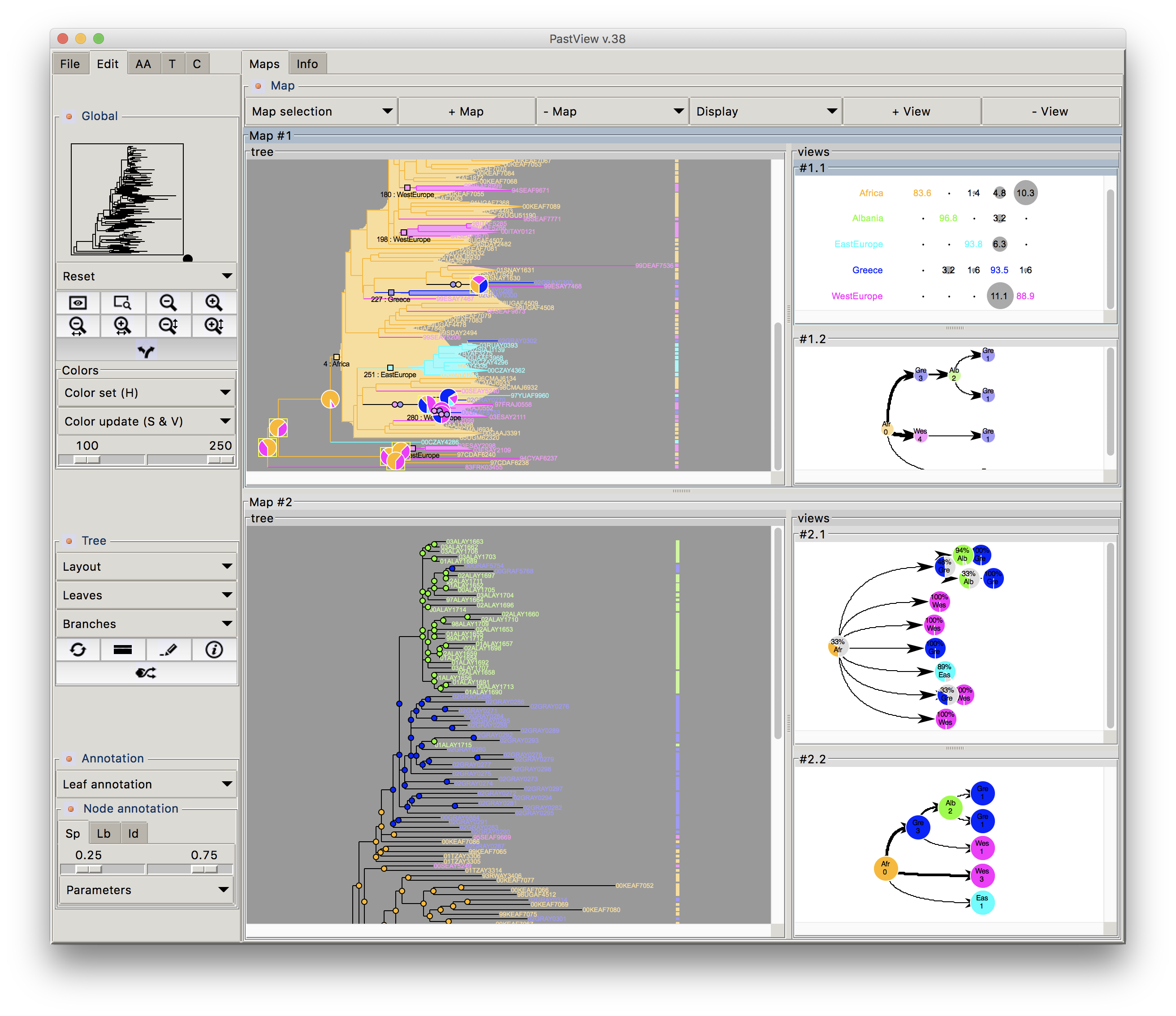
Phylogenetic trees are often combined with extrinsic traits to reconstruct evolutionary scenarios in various research fields, including phylogeography, molecular epidemiology, and ecology. Analysis starts with the computation of ancestral annotations from extrinsic traits (discrete variables such as geographic origin, resistance to a treatment, and life history traits), associated with the sampled sequences used to build the phylogenetic tree. Analysis continues with the study of the evolutionary changes from the tree root to its tips to characterize evolutionary scenarios such as the spread of a disease, the dynamic of a drug resistance, or shifts in ecological habitats. Difficulties may arise when interpreting the output. First, there is a diversity of optimization criteria for computing trees and ancestral annotations (parsimony, maximum likelihood, Bayesian) and each of them involves various models of evolution; these methods may all yield different results that need to be compared. Second, the tree size and complexity of the annotations can be an inconvenience with respect to computation time or tedious graphical analysis. Third, although probabilistic models give more accurate results than other methods, if some ancestral characters do not stand out clearly from others (i.e. they are much more likely), they may produce a combinatorial explosion of potential evolutionary scenarios. To the best of our knowledge, there are no tools to help with the comparison of different sets of ancestral annotations and find common patterns across multiple evolutionary scenarios from a beam of transitions. We thus present PastView, a new editor with a user-friendly interface that includes numerical and graphical features to help users in this context. PastView is available publicly as a standalone software. Contact : francois.chevenet@ird.fr
 Sep. 2019 PastView release (V46) (Bugs corrected for the Windows version)
Jul. 2019 PastView release (V45) & User's Manual 1.1 & Tutorial
May. 2019 PastView release (V43, new compression level for transitions maps) & PastView web interface
Nov. 2018 PastView release (V40, transitions matrices revisited)
Sep. 2018 PastView release (V39, transitions query revisited)
Jul. 2018 PastView first release
Sep. 2019 PastView release (V46) (Bugs corrected for the Windows version)
Jul. 2019 PastView release (V45) & User's Manual 1.1 & Tutorial
May. 2019 PastView release (V43, new compression level for transitions maps) & PastView web interface
Nov. 2018 PastView release (V40, transitions matrices revisited)
Sep. 2018 PastView release (V39, transitions query revisited)
Jul. 2018 PastView first release
|
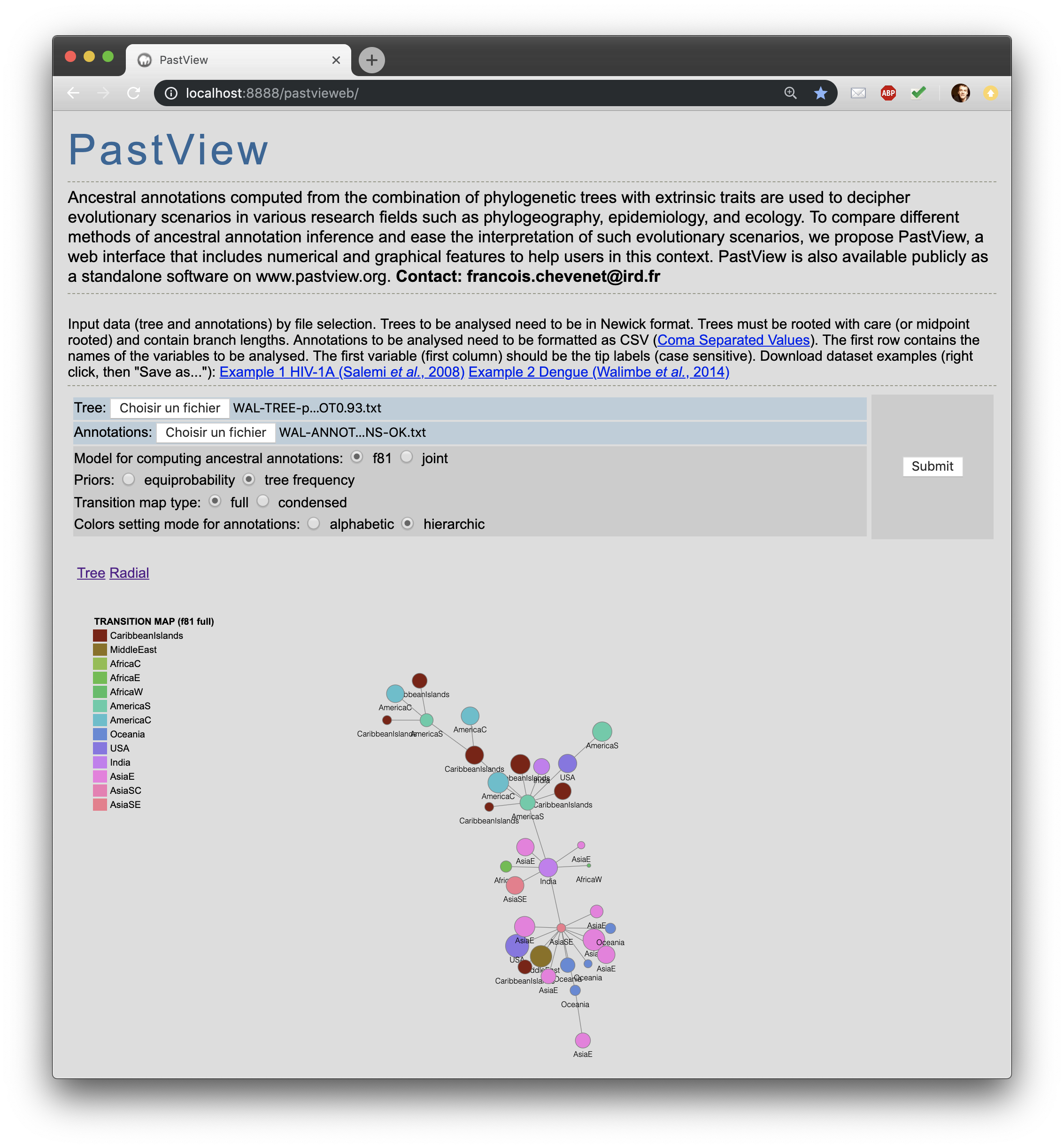
A PastView web interface is available here. It is still under construction and it will be updated ASAP. |
1) Download & Install Tcl/Tk: OSX Linux Windows
2) Download PastView package (source code, multi-platforms)
3) under Linux or OSX: from a terminal, goto to the downloaded PastView folder, then command "wish", then from the wish prompt, command "source pastview.tcl". Under Windows: double click the pastview.tcl file.
Documentation

|
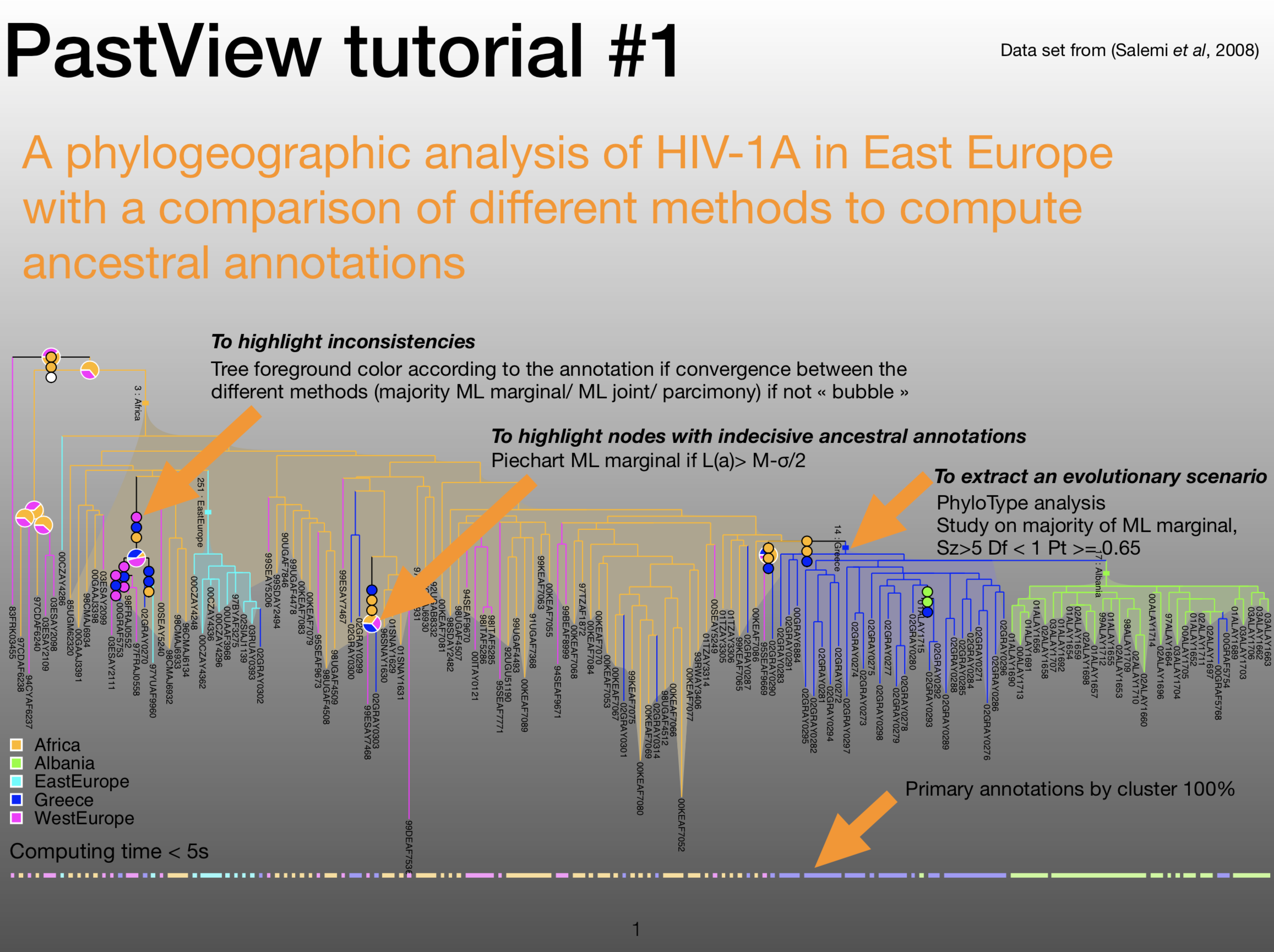
|
|
IRD - MIVEGEC (IBC) |
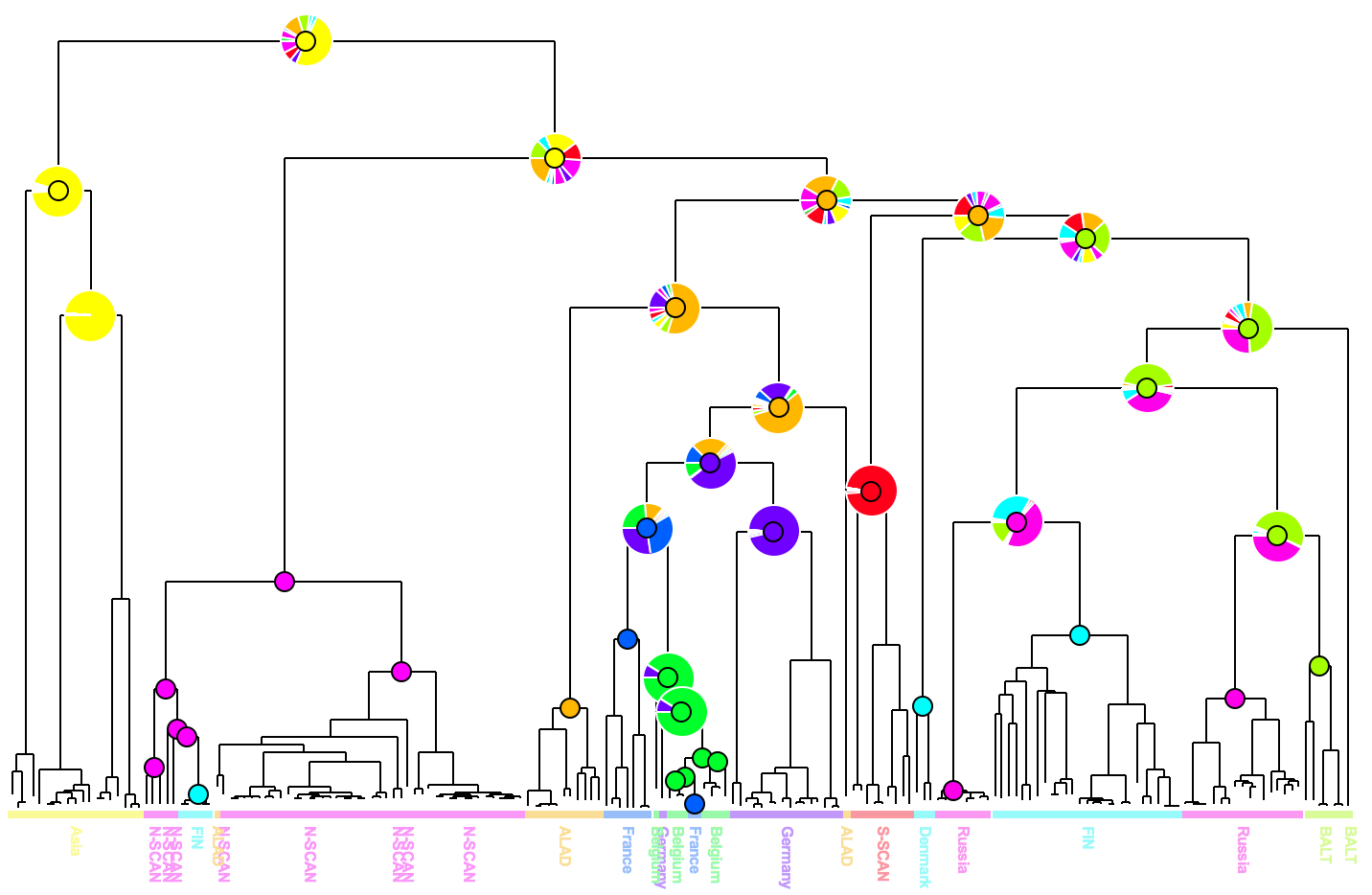
|